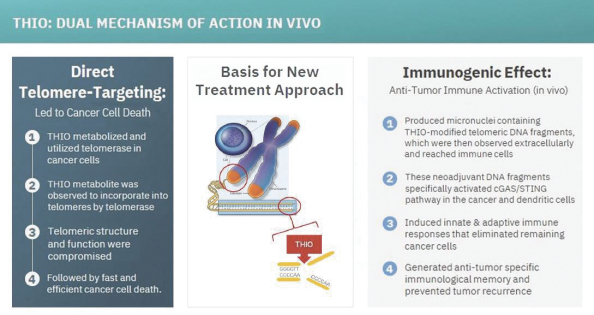
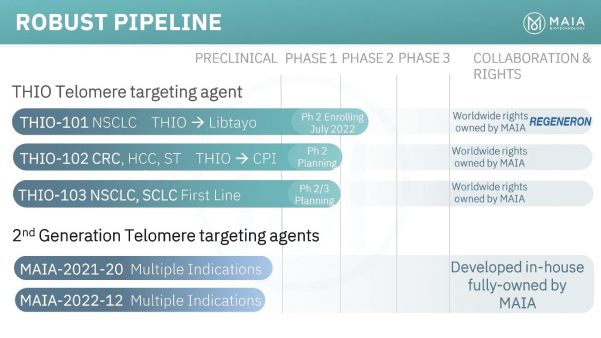
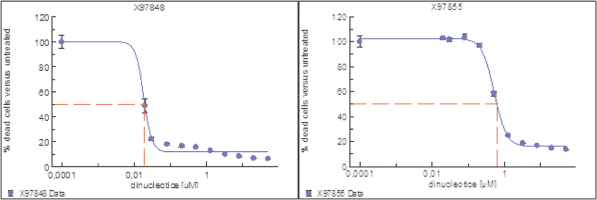
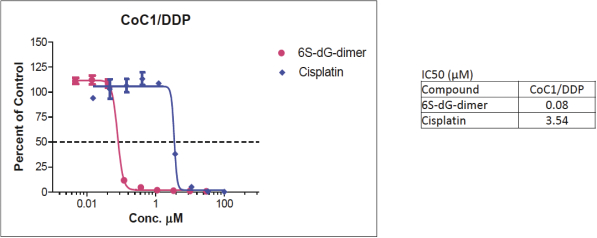
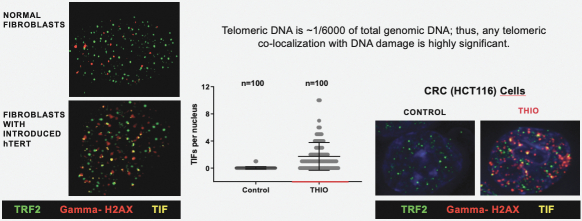
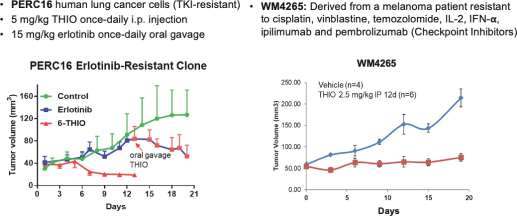
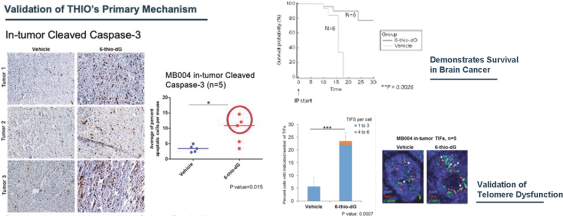
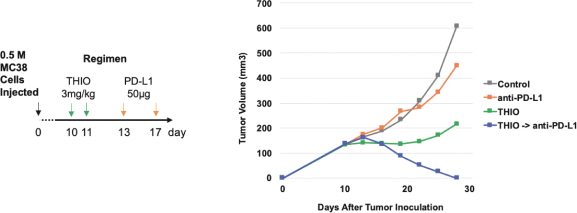
of their securities. This risk is especially relevant for us because biotechnology companies have experienced significant share price volatility in recent years. Securities litigation against us, regardless of the merits or outcome, could result in substantial costs and divert the time and attention of our management from our business, which could have a material adverse effect on our business, financial condition, and results of operations.
Future sales of our common stock, or the perception in the public markets that these sales may occur, could cause the market price for our common stock to decline.
All shares of common stock sold in this offering will be freely transferable without restriction or further registration under the Securities Act. At the time of this offering, 2,032,909 shares of common stock are reserved for issuance under our 2021 Equity Incentive Plan. We cannot predict the effect, if any, that market sales of shares of our common stock or the availability of shares of our common stock for sale will have on the market price of our common stock prevailing from time to time. Sales of substantial amounts of shares of our common stock in the public market, or the perception that those sales will occur, could cause the market price of our common stock to decline. Of the shares of common stock outstanding, 8,635,904 are restricted securities within the meaning of Rule 144 under the Securities Act and subject to certain restrictions on resale. Restricted securities may be sold in the public market only if they are registered under the Securities Act, or are sold pursuant to an exemption from registration such as Rule 144 or Rule 701.
We, each of our officers, directors, and certain of our stockholders agreed in connection with our initial public offering, which was consummated on August 1, 2022, subject to certain exceptions, not to offer, pledge, sell, contract to sell, grant, lend, or otherwise transfer or dispose of, directly or indirectly, or enter into any swap or other arrangement that transfers to another, in whole or in part, any of the economic consequences of ownership of any shares of our capital stock or any securities convertible into or exercisable or exchangeable for shares of our common stock, for a period of 12 months after the date of our IPO prospectus, without the prior written consent of the representative. Certain of our other stockholders, who are not insiders, have agreed, subject to certain exceptions, not offer, pledge, sell, contract to sell, grant, lend, or otherwise transfer or dispose of, directly or indirectly, or enter into any swap or other arrangement that transfers to another, in whole or in part, any of the economic consequences of ownership of any shares of our capital stock or any securities convertible into or exercisable or exchangeable for shares of our common stock, for a period of 12 months after the date of our IPO prospectus, without the prior written consent of the representative. In connection with the offering that closed on April 27, 2023, our directors and officers agreed, subject to certain exceptions, not to offer, pledge, sell, contract to sell, grant, lend, or otherwise transfer or dispose of, directly or indirectly, or enter into any swap or other arrangement that transfers to another, in whole or in part, any of the economic consequences of ownership of any shares of our capital stock or any securities convertible into or exercisable or exchangeable for shares of our common stock, until October 24, 2023. In connection with this offering, our directors and officers have agreed, subject to certain exceptions, not to offer, pledge, sell, contract to sell, grant, lend, or otherwise transfer or dispose of, directly or indirectly, or enter into any swap or other arrangement that transfers to another, in whole or in part, any of the economic consequences of ownership of any shares of our capital stock or any securities convertible into or exercisable or exchangeable for shares of our common stock, for a period of three (3) months from the date of this offering. Following the expiration of the applicable lock-up period, all of the issued and outstanding shares of our common stock will be eligible for future sale, subject to the applicable volume, manner of sale, holding period, and other limitations of Rule 144. See the section of this prospectus entitled “Underwriting” for additional information. See “Underwriting” for additional information. Following the expiration of the applicable lock-up period, all of the issued and outstanding shares of our common stock will be eligible for future sale, subject to the applicable volume, manner of sale, holding period, and other limitations of Rule 144.
If securities or industry analysts publish unfavorable research about our business, or if our competitors’ stock performance decline, the price of our common stock and our trading volume could decline.
The trading market for our common stock depends in part on the research and reports that securities or industry analysts may publish about us or our business. We do not have any control over these analysts. If one or more of
59